
El sistema nervioso se encarga de numerosas funciones en nuestro organismo, siendo capaz de coordinar un fino gesto, como el levantar el meñique, hasta planificar el regalo perfecto para un ser querido. Como ocurre con todo el cuerpo humano, es muy complejo, y por ello, para comprenderlo, tenemos que entender su componente más simple: la neurona, su unidad funcional.
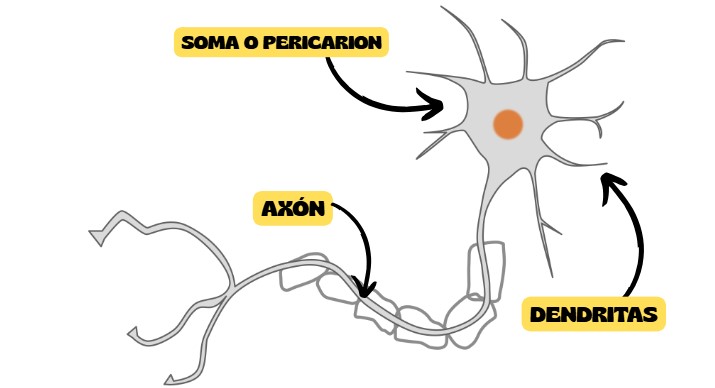
Las neuronas se componen principalmente de tres estructuras:
-El cuerpo celular (también denominado soma o pericarion), que contendrá el núcleo, así como la mayoría de los orgánulos celulares. Será el centro trófico y de síntesis de toda la neurona, por lo que el soma es esencial para mantener viva y funcional a la célula.
-Las dendritas, que son numerosos procesos elongados que se extienden desde el cuerpo celular y están especializados en recibir estímulos de otras neuronas a través de las denominadas “sinapsis”, puntos de conexión funcional entre neuronas que sirven para transmitir información.
-El axón, un único y largo proceso que se extiende desde el soma y que está especializado en generar y conducir impulsos nerviosos a otras células (pudiendo ser otras neuronas, a células musculares o a células glandulares). Los axones podrán recibir también información de otras neuronas, modificando así sus potenciales de acción. El axón se ramificará en terminales axónicos los cuales podrán formar sinapsis con las células mencionadas.
Además de las neuronas en el sistema nervioso, se encontrarán las células gliales que mantendrán un ambiente óptimo para las neuronas y su actividad. Hay seis células gliales principales: oligodendrocitos (encargados de mielinización de axones en el sistema nervioso central), las células de Schwann (encargadas de la mielinización de axones en el sistema nervioso periférico), los astrocitos (responsables del mantenimiento estructural y metabólico de las neuronas), las células ependimarias (ayudan en la producción y el movimiento del líquido cefalorraquídeo), la microglía (pertenecientes a la familia de los macrófagos serán encargados de la defensa de las neuronas y de su inmunidad) y, por último, las células satélite (darán soporte estructural y metabólico a los cuerpos neuronales). (Mescher, 2018)
Dado que la mayor concentración de neuronas se encuentra en el cerebro, cualquier alteración en su estructura o función puede dar lugar a diversas patologías neurológicas y que derivarán en problemas, entre ellos cognitivos, afectando diferentes funciones como el habla, la memoria, la personalidad, etc. La demencia es un síndrome caracterizado por la pérdida de la memoria reciente y de otras funciones intelectuales cuyo inicio suele ser insidioso, pero progresa constantemente. La enfermedad del Alzheimer es la demencia más frecuente y constituye el 60-80% de los casos en los ancianos. (Purves et al., 2012)
En los casos típicos del Alzheimer, el primer signo será un deterioro de la memoria reciente y de la atención que continuará con el posterior deterioro del juicio, la orientación visoespacial, el pensamiento abstracto y el lenguaje. Esto conducirá a alteraciones en la personalidad del sujeto que lo padezca. El diagnóstico de esta demencia sólo se podrá confirmar mediante la patología celular definida, en el examen post mortem del encéfalo.
La histopatología presentará tres rasgos predominantes:
-Acumulación de placas amiloides en el espacio extracelular. Esta acumulación se producirá por diferentes causas según la forma de la enfermedad sea temprana o tardía.
–Ovillos neurofibrilares en el citoplasma de las neuronas. Se producen por la hiperfosforilación de las proteínas tau. La acumulación de las placas de β amiloide provocará una cascada de cinasas causando la fosforilación de las proteínas, así como un estrés oxidativo también clave en la formación de estos ovillos neurofibrilares. Las alteraciones en la función estabilizadora de la proteína tau conducen a la acumulación de pares retorcidos de tau dentro de las neuronas. En las neuronas normales, la tau soluble favorece el ensamblaje y la estabilidad de los microtúbulos, así como el transporte vesicular axonal. La tau hiperfosforilada es insoluble, pierde afinidad por los microtúbulos y se autoasocia en filamentos helicoidales pareados. (Abraham L. Kierszenbaum, 2020)
–Pérdida difusa de neuronas.
La mayoría de los casos de la enfermedad del Alzheimer aparecen esporádicamente y de forma tardía después de los 60 años. Se deberá al envejecimiento, principalmente (manteniendo importancia también el estilo de vida), y a la pérdida de capacidad de eliminar acumulación de proteínas, lo que lleva a la formación de los depósitos de placa amiloides que posteriormente contribuirán a la generación de los ovillos neurofibrilares debido al estrés oxidativo y a la cascada de cinasas. Sin embargo, también se ha descubierto que el heredar genéticamente el alelo ε4 en la Apolipoproteína E será un factor de riesgo en esta enfermedad, aumentando la probabilidad de su desarrollo en el portador de este gen. (Purves et al., 2012)

Por el contrario, puede darse esta enfermedad en la mediana edad (entre los 30 y 60 años mayormente). Esto es relativamente raro y se debe a defectos monogénicos compatibles con un patrón autosómico dominante. Estarán involucrados en la enfermedad la mutación de los genes que codifican la proteína precursora del amiloide (APP). Este gen APP se identificaría mutado en algunos pacientes con la enfermedad de inicio temprano y ninguno de los que la presentan de forma tardía. Posteriormente también se hallarían genes mutantes que subyacen a otras dos formas autosómicas dominantes adicionales de enfermedad de Alzheimer (presenilina 1 y prenisilina 2). Las mutaciones de estos dos genes alteran el procesamiento de la APP conduciendo a cantidades mayores de una forma particularmente tóxica del péptido Aβ, Aβ42. La mutación de cualquiera de estos genes parece ser suficiente para dar lugar al desarrollo de una forma hereditaria de Alzheimer, convergiendo estos en el anormal procesamiento de la APP. (Purves et al., 2012)
Indudablemente, la enfermedad de Alzheimer tiene una anatomía patológica compleja que es probable que refleje distintas anomalías moleculares y celulares relacionadas. Hasta ahora, el común denominador más evidente en esta enfermedad compleja es el procesamiento anormal de la proteína precursora del amiloide. En particular, se cree que la acumulación del péptido Aβ42 tóxico es un factor clave. Esta conclusión ha conducido a esfuerzos por desarrollar terapias dirigidas a inhibir la formación o facilitar la depuración de este péptido tóxico. (Purves et al., 2018)
Un ejemplo de estas terapias es lecanemab, un anticuerpo monoclonal humanizado diseñado para unirse específicamente a los protofibrilos solubles de Aβ42. Estudios post-mortem han demostrado que lecanemab se une a placas amiloides difusas y compactas, así como a Aβ intraneuronal, sin afectar fibrillas vasculares ni monómeros, lo que sugiere una acción terapéutica selectiva. Esta unión facilita la eliminación de amiloide mediante activación de la microglía, promoviendo su depuración y reduciendo la carga amiloide cerebral, lo cual podría ralentizar el deterioro cognitivo observado en pacientes con enfermedad de Alzheimer temprana. (Johannesson et al., 2024)
Bibliografía:
Johannesson, M., Söderberg, L., Zachrisson, O., Fritz, N., Kylefjord, H., Gkanatsiou, E., Button, E., Svensson, A. S., Rachalski, A., Nygren, P., Osswald, G., Lannfelt, L., & Möller, C. (2024). Lecanemab demonstrates highly selective binding to Aβ protofibrils isolated from Alzheimer’s disease brains. Molecular and cellular neurosciences, 130, 103949. https://doi.org/10.1016/j.mcn.2024.103949
Kierszenbaum, A. L., & Tres, L. L. (2020). Histology and cell biology: An introduction to pathology (5ª ed.). Elsevier.
Mescher, A. L. (2018). Junqueira’s basic histology: Text and atlas (15ª ed.). McGraw-Hill Education.
Purves, D., Augustine, G. J., Fitzpatrick, D., Hall, W. C., LaMantia, A.-S., & White, L. E. (2012). Neurociencia (5ª ed.). Editorial Médica Panamericana.
Purves, D., Augustine, G. J., Fitzpatrick, D., Hall, W. C., LaMantia, A.-S., & White, L. E. (2018). Neuroscience (6ª ed.). Sinauer Associates


